Overview of regtools variants annotate command
The regtools variants annotate command is used to annotate variants of interest with annotations of interest. For example to annotate variants that might fall in regions that affect the splicing machinery.
Usage
regtools variants annotate [options] variants.vcf annotations.gtf annotated_output.vcf
Input
| Input | Description |
|---|---|
| variants.vcf | The variants to be annotated in the VCF format. The htslib VCF parser prefers all chromosome names be present as contig lines in the header. If this is not the case, please tabix index the file. Regtools accepts gzipped VCF files, i.e *.vcf.gz |
| annotations.gtf | The GTF file specifies the transcriptome that is used to annotate the variants. For examples, the Ensembl GTFs for release78 are here |
Options
| Option | Description |
|---|---|
| -i | Maximum distance from the start/end of an exon to annotate a variant as relevant to splicing, the variant is in intronic space. [default = 2] |
| -e | Maximum distance from the start/end of an exon to annotate a variant as relevant to splicing, the variant is in exonic space, i.e a coding variant. [default = 3] |
| -I | Annotate variants in intronic space within a transcript (not to be used with -i). |
| -E | Annotate variants in exonic space within a transcript (not to be used with -e). |
| -S | Dont skip single exon transcripts. The default is to skip the single exon transcripts. |
| -o | Name of output file, this file will be in the VCF format. [STDOUT] |
Output
The output file is in the VCF format. The annotation results are described using fields in the INFO column.
| INFO column field | Description |
|---|---|
| genes | A comma separated list of unique genes that the variants falls in. |
| transcripts | A comma separated list of transcripts that the variants falls in. |
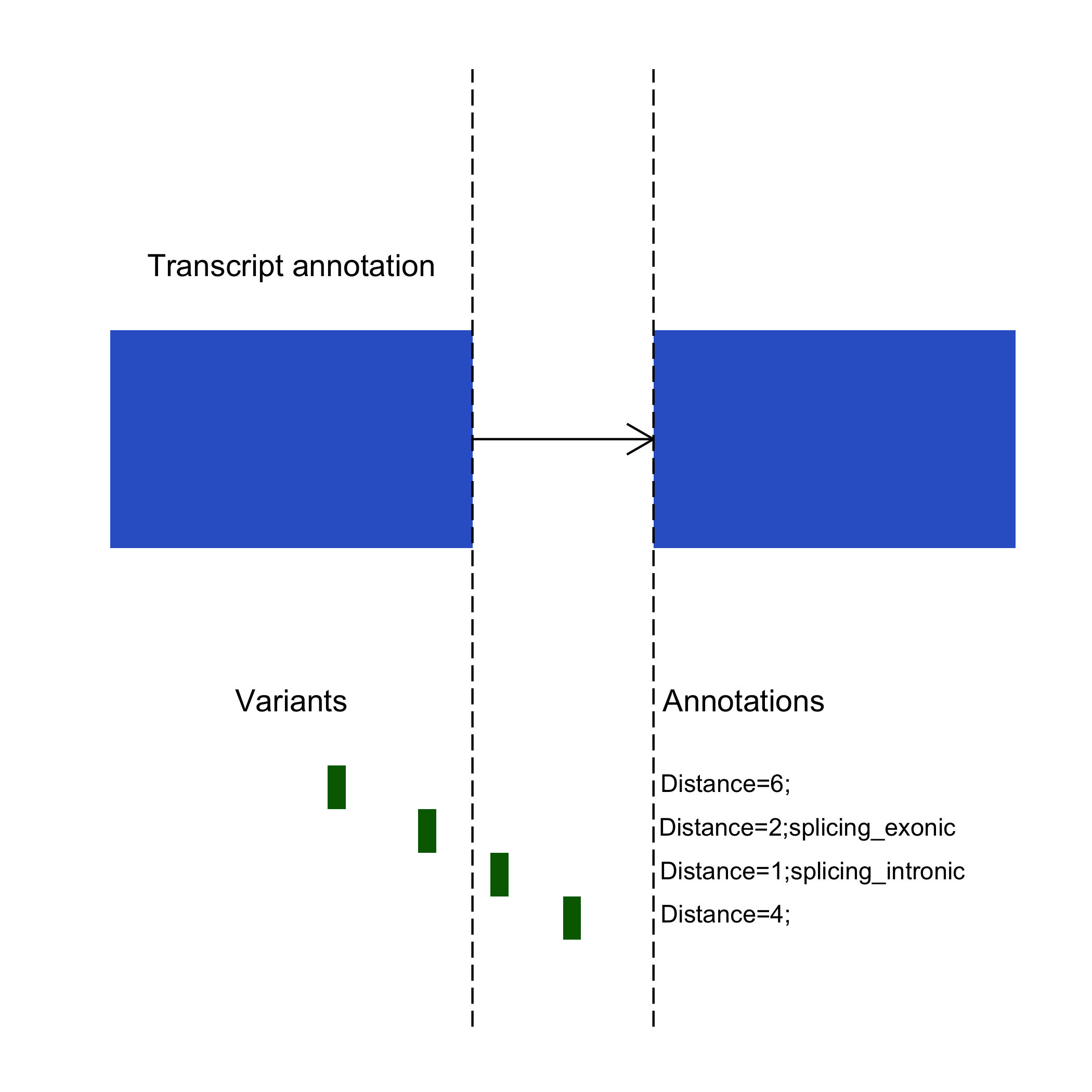
| distances | A comma separated list of distances from the start or stop of an exon. This distance is min(distance_from_start_of_exon, distance_from_end_of_exon). The number of elements in the list is same as the number of transcripts in the 'transcripts' field and in the same order. The distance needs to be less than or equal to the parameters specified by the -i and -e options. |
| annotations | A comma separated list of annotations in the same order as the 'transcripts'. The number of elements in this list is the same as the number of transcripts in the 'transcripts' field and in the same order, i.e each annotation corresponds to a different transcript. The valid values for this field are 'splicing_exonic' and 'splicing_intronic'. If the variant lies within the distance specified by '-e' option (3 b.p by default) inside an exon, it is annotated as 'splicing_exonic'. If the variant lies outside the exon within the distance specified by the '-i' option (2 b.p by default), it is annotated as 'splicing_intronic'. Note that even when all intronic or exonic variants are considered with the -I or -E options, they are labelled simply as "intronic" or "exonic", respectively. |
Example output line
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT A B
22 175311 . G T 29.2 PASS AN=4;AC=2;genes=RANGAP1;transcripts=ENST00000356244,ENST00000405486,ENST00000407260,ENST00000455915;distances=2,2,2,2;annotations=splicing_intronic,splicing_intronic,splicing_intronic,splicing_intronic GT:GQ 0/1:215 0/1:225
Example annotation