Overview of regtools junctions annotate command
The regtools junctions annotate command is a tool to annotate the observed junctions with respect to a known
transcript structure. The known transcript structure is in the form of a GTF file obtained from one of the standard
Gene Annotation databases such as Ensembl/RefSeq/UCSC etc. The goal of the annotation step is to help identify novel/unusual junctions.
Usage
regtools junctions annotate [options] junctions.bed ref.fa annotations.gtf
Input
| Input | Description |
|---|---|
| junctions.bed | The BED file with the junctions that have be annotated. This file has to be in the BED12 format. One recommended way of obtaining this file is by running regtools junctions extract. See here for more details. |
| ref.fa | The reference FASTA file. The donor and acceptor sequences used in the "splice-site" column are extracted from the FASTA file. |
| annotations.gtf | The GTF file specifies the transcriptome that is used to annotate the junctions. For examples, the Ensembl GTFs for release 106 are here. |
Options
| Option | Description |
|---|---|
| -S | Do not skip single exon genes. The default is to skip the single exon genes while annotating junctions. |
| -o | File to write output to. STDOUT by default. The output format is described here |
| -h | Display help message for this command. |
Output
| Column name | Description |
|---|---|
| chrom | Chromosome of the junction. |
| start | Junction start co-ordinate. [zero based format] |
| end | Junction end co-ordinate. [zero based format] |
| name | Identifier for the junction. |
| score | The number of reads supporting the junction. [integer] |
| strand | The strand the junction is identified. Same as the input file. [+/-] |
| splice_site | The two basepairs at the donor and acceptor sites separated by a hyphen. [e.g CT-AG] |
| acceptors_skipped | Number of known acceptors skipped by this junction according to the GTF. See Notes below for explanation. [integer] |
| exons_skipped | Number of known exons skipped by this junction according to the GTF. See Notes below for explanation. [integer] |
| donors_skipped | Number of known donors skipped by this junction according to the GTF. See Notes below for explanation. [integer] |
| anchor | Field that specifies the donor, acceptor configuration. See Notes below for explanation. [D/A/DA/NDA/N] |
| known_donor | Is the junction-donor a known donor in the GTF file? [0/1] |
| known_acceptor | Is junction-donor a known acceptor in the GTF file? [0/1] |
| known_junction | Does the junction have a known donor-acceptor pair according to the GTF file. This is equivalent to "DA" in the "anchor" column. |
| transcripts | The transcripts that overlap the junction according to the input GTF file. |
| genes | The genes that overlap the junction according to the input GTF file. |
Notes
Annotating observed junctions with known donor/acceptor/junction information
It is useful to annotate the ends of junction with respect to known acceptors, donors and junctions in the transcriptome. The known acceptor, donor and junction information is computed from the GTF file and this information is then used to annotate the observed junctions.
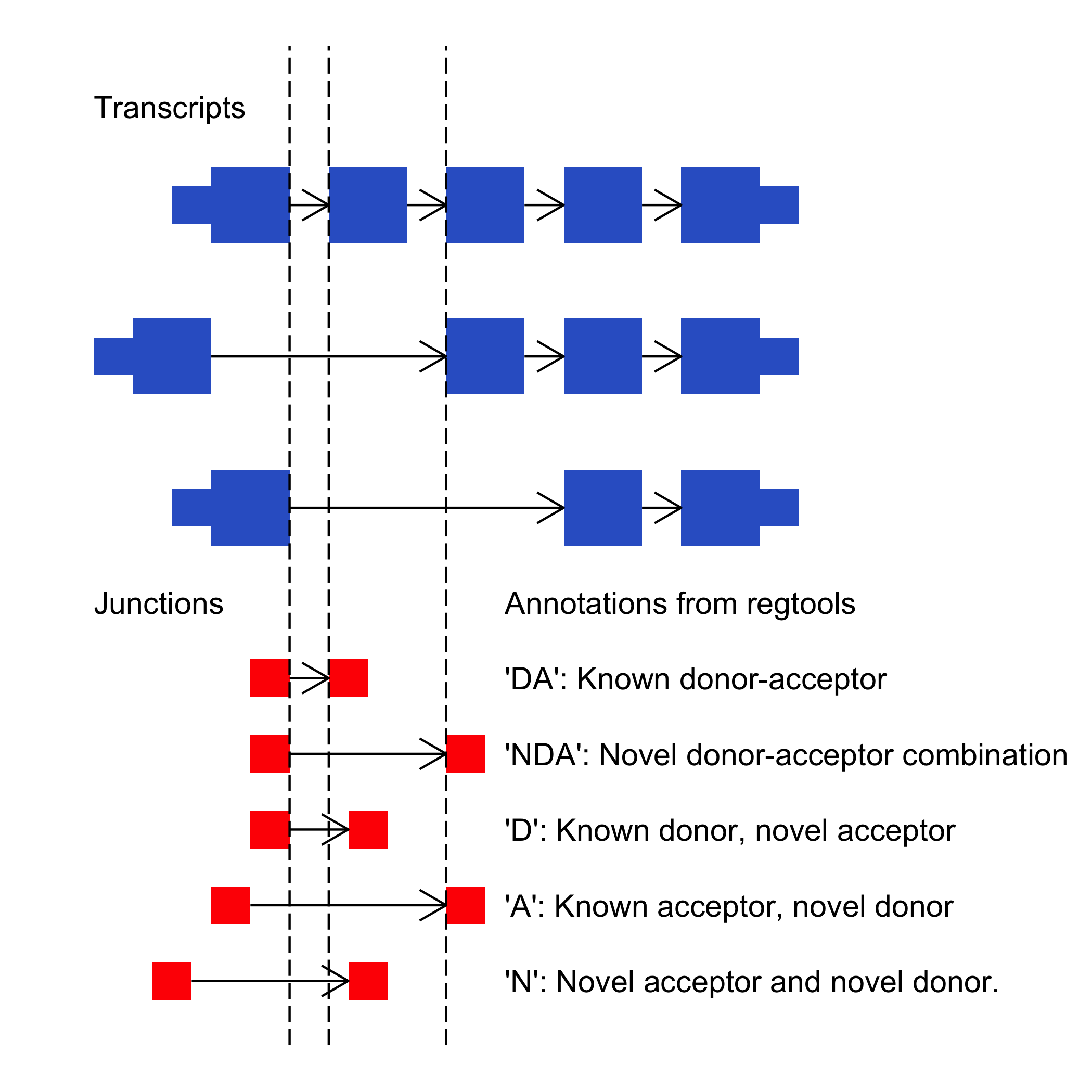
The junctions are annotated using the following nomenclature (and as shown in the figure below.)
-
DA - The ends of this junction are known donor and known acceptor sites according to "annotations.gtf". This junction is known to the transcriptome.
-
NDA - The ends of this junction are known donor and known acceptor sites, according to "annotations.gtf". This junction is not known to the transcriptome (novel).
-
D - The ends of this junction are a known donor site and a novel acceptor site, according to "annotations.gtf". This junction is not known to the transcriptome (novel).
-
A - The ends of this junction are a novel donor site and a known acceptor site, according to "annotations.gtf". This junction is not known to the transcriptome (novel).
-
N - The ends of this junction are a novel donor site and a novel acceptor site, according to "annotations.gtf". This junction is not known to the transcriptome (novel).
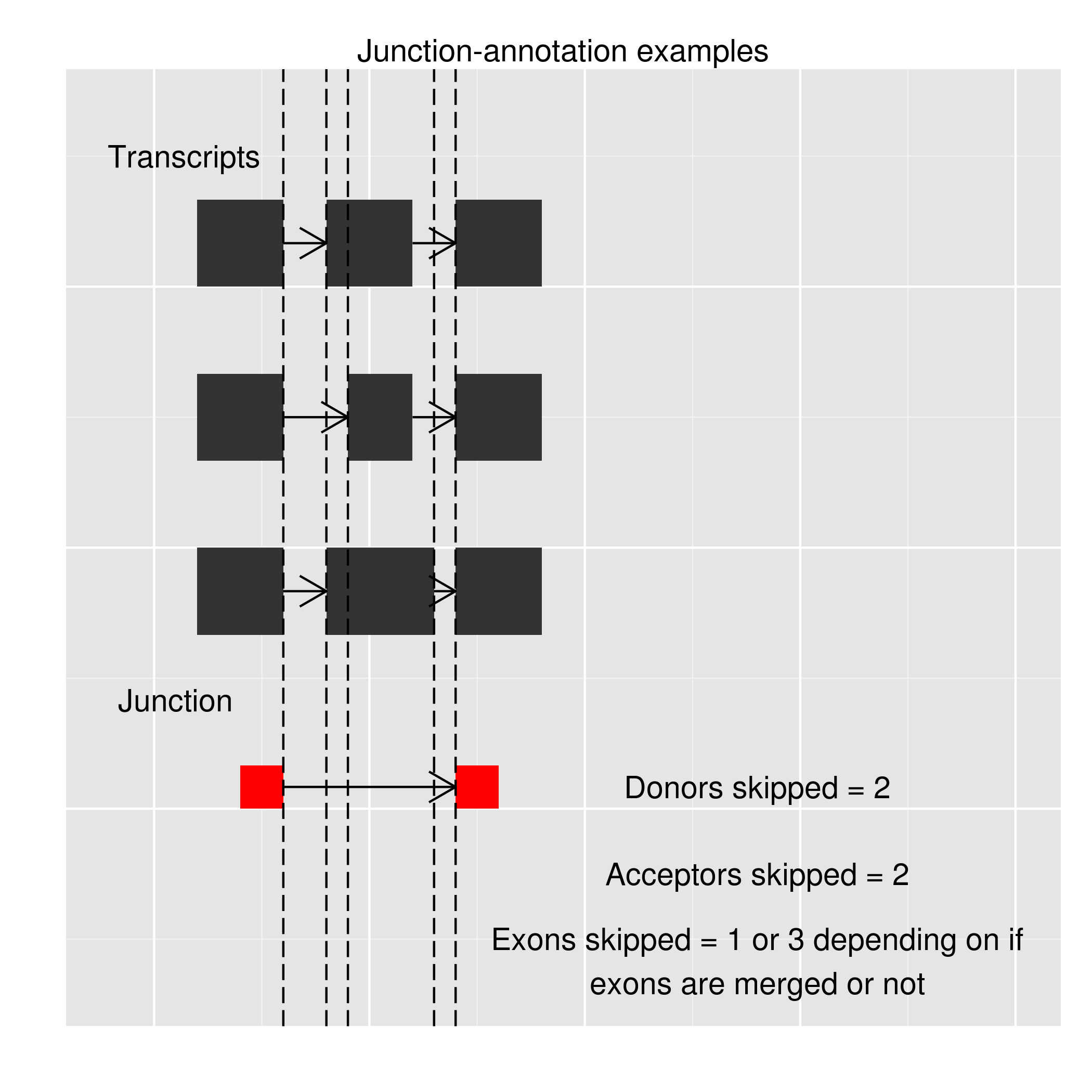
Annotating a junction with number of donors/acceptors/exons skipped
Exon skipping is a form of RNA splicing that can be identified using RNAseq data. It is hence useful to compute for every observed putative exon-exon junction, the number of exons skipped, the number of known donor sites skipped and the number of known acceptor sites skipped. The known exons, donors and acceptors are calculated from the user supplied GTF file.
In the example shown below, the observed junction has skipped 2 known donor sites and 2 known acceptor sites. For the number of exons skipped we consider two situations. The second exon in all the three transcripts overlap, if these overlapping exons are merged together, the number of exons skipped is just 1. If these three exons are considered to be different the number of exons skipped is 3. We try and provide both these annotations.
If any of the examples are not clear or if you would like more information please feel free to open an issue on GitHub here or post on the discussion page here.